How to bring down the price of drugs such as the novel coronavirus therapy remdesivir
Overview
The extraordinary ongoing effort to find treatments to cure, ameliorate symptoms of, and vaccinate against the novel coronavirus and COVID-19, the disease caused by the virus, is a reminder of how much the U.S. healthcare system, the U.S. economy, and we as individuals and families rely on these products, as well as the public research that goes into discovering them and the pharmaceutical companies that develop and manufacture them. This issue brief discusses the ownership and costs of prescription drugs, as well as potential policy initiatives in this area, through the lens of one prescription medication, the antiviral drug remdesivir.
Remdesivir is important because it became the first signal of hope amid the coronavirus pandemic, when research published in April 2020 suggested that it reduced the duration of symptoms for patients with severe COVID-19 infections by about 4 days on average, although it did not have a significant effect on mortality. Those results led the U.S. Food and Drug Administration to permit its use under an emergency use authorization, and for months, it was the only drug available with at least some proven real clinical benefits for COVID-19. On October 22, the agency formally approved remdesivir for the treatment of COVID-19 requiring hospitalization.
Approval by the Food and Drug Administration, however, means that a drug has some benefit but it does not guarantee any particular level of benefit. Indeed, in a recent study, we found that only about one-third of new drugs approved by the agency over the past decade were rated as having high therapeutic value. For remdesivir, recent data have shaken the foundations of its therapeutic potential. According to a pre-print of a major World Health Organization solidarity study from October 2020, remdesivir does not appear to affect patients’ mortality or their hospitalization duration, and it is not clear whether its effects on symptoms are additive to dexamethasone, another drug shown to work for severe COVID-19. Remdesivir still seems to display some anti-COVID-19 activity and may have a role in physicians’ therapeutic armamentarium, but what that role will be will continue to evolve as further data emerge.
Gilead Sciences Inc. owns all the relevant patents for remdesivir. After the drug was authorized for use against COVID-19, the company announced plans to donate 1.5 million doses. But that commitment will not meet demand, and the company announced in June a list price of more than $3,000 for a full treatment regimen. At that price, Gilead is reportedly on track to make more than $9 billion in revenue on remdesivir in 2020 and 2021 alone. With FDA approval in hand, the company will be able to set whatever price it wants for the drug for likely more than a decade.
Remdesivir, then, offers a good reminder of the need for policies that can achieve continuing prescription drug innovation, while also maintaining a fair price for U.S. patients and the health care system more broadly. This issue brief will provide a short history of the development of remdesivir, summarize the intellectual property laws governing prescription drugs in general and remdesivir in particular, and present policy options to affect the pricing of remdesivir and drugs like it going forward.
Briefly, the policy recommendations detailed below are divided among four basic stages of drug development, approval, and production:
- Stage 1: the discovery process, leading up to approval by the U.S. Food and Drug Administration
- Stage 2: the 12- to 14-year period of market exclusivity for brand-name drugs
- Stage 3: the transition to a competitive market with the introduction of generic versions of the medication
- Stage 4: the period when multiple generic versions of the drug can be on the market
Remdesivir has just completed stage 1. Other potential treatments for COVID-19, such as monoclonal antibodies, are at even earlier stages. While these policy recommendations represent an ambitious agenda with significant implications for a broad range of drugs, the crisis brought on by the coronavirus pandemic makes clear that such an agenda not only is necessary but can and should be considered in the near future.
In short, now is the best time to put many of these provisions in place, before remdesivir and other treatments and vaccines make further progress along the approval path.
Let’s turn first to the history of the development of remdesivir to demonstrate the clear role of public financing in the discovery and development of the drug, and its public policy implications.
The discovery and development of remdesivir
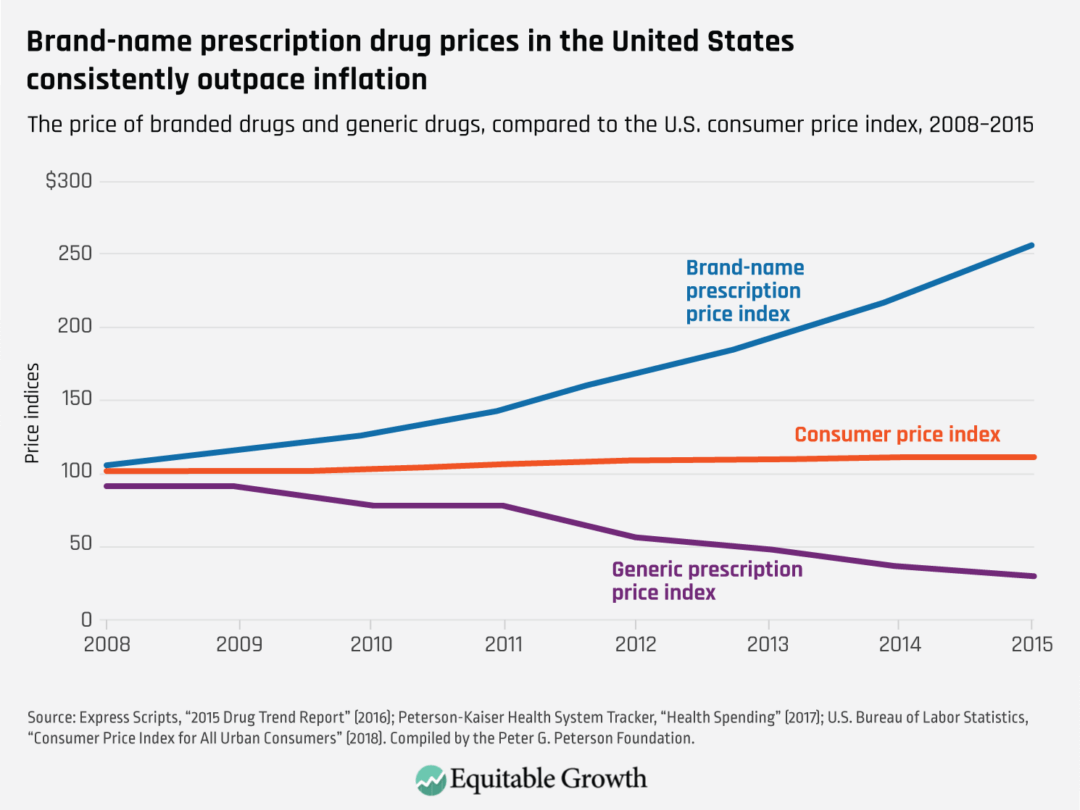
Prescription drugs can be very costly, which affects not only U.S. healthcare and the economy broadly but also individual health outcomes. High prices can keep important medications out of the hands of patients who need them to get well, or even to live. The price of brand-name prescription drugs has risen markedly faster than overall inflation over the past two decades. (See Figure 1.)
Figure 1
The federal government and not-for-profit organizations, especially U.S. universities—funded in good part by federal agencies—support or conduct most of the basic and translational research that lays the foundation for the medications we use. The role of public funding is more pronounced among the most transformative new drugs. The prescription drug industry also invests a great deal in bringing brand-name products to market—usually at the clinical testing, regulatory approval, and manufacturing phases of development—but the prices they charge are not related to the costs they incur in developing medications.
Yet U.S. patent laws give what is essentially monopoly pricing power to companies that hold the intellectual property related to valuable drugs, even if that intellectual property is derived from insights originating in the basic and translational research completed by publicly funded institutions and their scientists. And these firms sometimes use that power to charge prices far beyond the value that the drug provides. Given the federal government’s critical role, and the vital public interest in supporting public health, there are a number of concrete steps U.S. policymakers could take to better manage prescription drug costs at every stage of the approval process for these medications—without stifling innovation.
Remdesivir is a potent case in point.
The road that led to remdesivir’s development so that it could be effective against COVID-19 was paved by public funding and university research. Its initial development was the culmination of several years of collaboration between Gilead Sciences, the U.S. Army Medical Research Institute of Infectious Diseases, and the U.S. Centers for Disease Control and Prevention. The company and the two federal agencies were already collaborating in search of possible antiviral candidates in Gilead’s library of nucleoside analogues when the Ebola virus broke out in West Africa in 2014. The initiative identified the precursor to remdesivir, which company researchers and the government, led by the Army’s infectious diseases institute, further developed.
Researchers at Gilead Sciences and universities then turned to studying remdesivir as a potential treatment for various viruses, including coronaviruses, supported in part by another U.S. Department of Defense research agency. This joint public-private research involved groups such as the National Institutes of Health, that included the University of Alabama, the University of North Carolina, Chapel Hill, and Vanderbilt University. This research led to the discovery that remdesivir could be useful against Middle East Respiratory Syndrome, or MERS, and severe acute respiratory syndrome, or SARS.
Eventually, research conducted during the Ebola outbreak, sponsored by the government of the Democratic Republic of Congo and the African Coalition for Epidemic Research, Response, and Training, determined that remdesivir was not sufficiently effective against Ebola. But remdesivir was one of the earlier products tested against COVID-19, based on its usefulness against other coronaviruses causing MERS and SARS. Clinical trials were launched around the world beginning in early 2020 to evaluate the safety and efficacy of remdesivir in hospitalized patients with severe cases of the disease.
After several promising but insufficient trials, the National Institute of Allergies and Infectious Diseases, or NIAID, initiated the first double-blind randomized U.S. trial of remdesivir in February 2020. The trials showed that the drug could reduce recovery time by 4 days but did not lead to a significant reduction in deaths. It was not a gamechanger, but it was effective—and was the first drug identified to have a palliative effect on COVID-19.
Intellectual property and remdesivir
The results of remdesivir’s NIAID trial, along with other supporting data, led to its emergency use authorization and Gilead Sciences’ announcement in May that it would provide a set number of free doses of remdesivir, and then to its subsequent price announcement. Gilead set a price, however, that raised important concerns about the cost and availability of what was then the primary useful drug against COVID-19. Indeed, a number of state attorneys general urged federal action to address these issues, and state treasurers are calling on Gilead Sciences to lower the drug’s price. These calls have become more compelling with the recent data from the large multinational trial suggesting that the drug does not have benefits in terms of mortality or hospitalization.
Now that the drug is FDA-approved, Gilead remains in control of manufacturing, pricing, and distribution until its patents and other statutory exclusivities expire, and generic versions are approved by the Food and Drug Administration. There are several laws that provide exclusivity to manufacturers, but two that apply specifically to new medicines are especially relevant when discussing remdesivir. The Drug Price Competition and Patent Term Restoration Act of 1984—the Hatch-Waxman Act—gives drug companies a minimum of 5 years to 7 years of exclusivity for new drugs. Yet most drug manufacturers have patents lasting much longer than that. In fact, Gilead currently owns at least 12 patents on remdesivir. The last of them does not expire until 2039. But such patents can sometimes be challenged or, if necessary, designed around to enable generic drugs to enter the market long before patents expire. The Hatch-Waxman Act created a pathway for such challenges to occur, and it created a mechanism for generic manufacturers to seek FDA approval when the patents expire.
Another law offering exclusivity at the time of drug approval is the Orphan Drug Act, enacted in 1983, which provides 7 years of exclusivity and a valuable tax credit for drugs used in treating rare diseases (those affecting fewer than 200,000 people). The purpose of this law was to encourage pharmaceutical companies to develop drugs for rare diseases that might not be profitable, given the limited market for them. Early in the COVID-19 pandemic, when the number of infections was below 200,000, Gilead Sciences requested Orphan Drug Act designation for remdesivir for the treatment of COVID-19. The Food and Drug Administration approved this status in March, despite the rising numbers of cases—until public scrutiny led the company to withdraw this designation voluntarily.
Public financial support for development of remdesivir
As noted above, remdesivir, like most drugs, stands on the shoulders of federally funded research. In some respects, that is truer for this drug than for many others, given the specific partnerships formed between federal agencies and Gilead Sciences, culminating with the recognition that the drug might be useful for COVID-19. (See Table 1.)
Table 1
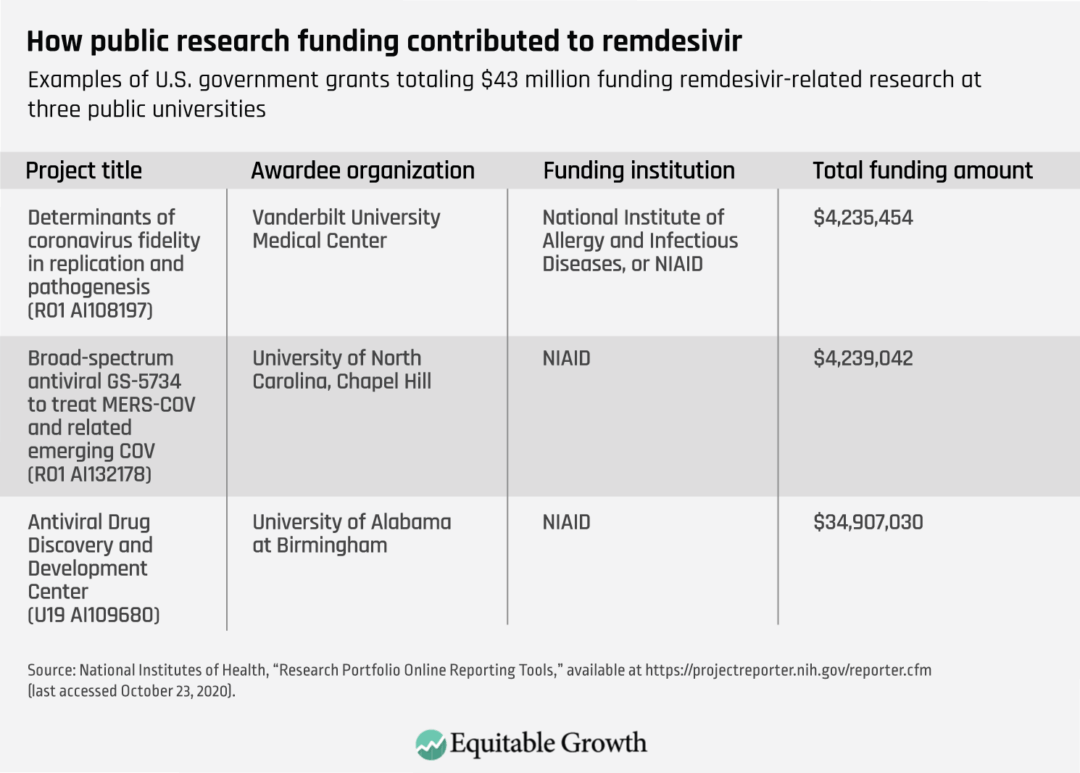
U.S. government research laboratories and university research funded by the federal government—mainly the National Institutes of Health—are the origin of many fundamental discoveries on which new drugs are based. Drug companies then spend a considerable amount developing and bringing a product to market. This is generally accomplished by the pharmaceutical company that comes to own the intellectual property for a given compound. Yet companies charge high prices to the very taxpayers who funded the research that made their product possible, and do not direct proceeds to the federal government to help fund new research. In the case of remdesivir amid the coronavirus recession, these considerations are now front and center for policymakers.
Policy options
There are numerous legislative and regulatory actions that policymakers could take to address drug prices that have risen far beyond inflation in the past three decades. All of them taken together would significantly lower spending on prescription drugs while ensuring continued funding for true innovation. Applying such measures to the case of remdesivir can be further justified by the federal funding that provides all or part of the foundation for the advances subsequently funded by Gilead. (For a more thorough description of these ideas, which address problems in four key periods of the development of pharmaceuticals, see my essay in Equitable Growth’s Vision 2020: Evidence for a stronger economy.)
Drug discovery period
When it supports research that could lead to the discovery or understanding of prescription drugs, the National Institutes of Health could require a reasonable pricing provision to be attached to the provision of funding. This provision could, for example, require that the ultimate price of the product be no greater than its value-based price—a price reflecting the drug’s potential ability to improve patient outcomes over comparable interventions—as determined by independent organizations.
In the case of remdesivir, the Institute for Clinical and Economic Review used cost-effectiveness analysis to judge that a fair price for remdesivir initially would be $4,460, assuming it had an effect on mortality, which was not statistically significant in the initial trial. Without an effect on mortality, as was confirmed by the more recent multinational trial, the institute estimated that a cost-effective price of remdesivir would be just $310 for a course of therapy.
Another option—not relevant in the case of remdesivir—would be for the National Institutes of Health to change its interpretation of the “march-in” provision of the Bayh-Dole Act of 1980, which established the basic rules for commercialization of technology arising from government funding. Under Bayh-Dole, the NIH retains a license on patents resulting from federally funded research and can “march in” to invalidate an exclusive commercialization license if the product is not made available on “reasonable terms.” The institute has never applied this provision to pricing matters, but this can be a tool for curbing high prices in a limited number of cases. This provision would not be relevant in the case of remdesivir because the government does not have a direct stake in the patents held by Gilead.
Brand-name-only period
Until the availability of generic substitutes for remdesivir, there will be no direct competition to bring down prices. Introduction of other brand-name products indicated for the same purpose have generally not been shown to lower prices to a substantial extent. Until generic competition, then, the best way to achieve fair prices for brand-name drugs is to empower the buyers to negotiate better terms with the manufacturers. There is no larger U.S. buyer than the 44 million Americans covered by Medicare.
Medicare, however, is barred from negotiating drug prices—unlike the U.S. Department of Veterans Affairs—a prohibition that does not apply to any other health care service it covers. So, the best solution for managing drug prices during this period is to provide the entire federal government, including Medicare, the authority to negotiate reasonable prescription drug prices that better reflect the value of treatments, as well as the government’s budget and the drugs’ origins.
To accomplish this, the United States could follow the model established by numerous other countries. U.S. policymakers could create a health technology assessment organization that would evaluate a newly approved drug’s clinical value and help determine a fair price based on how well it is expected to perform against other available treatments as well as other relevant factors. That assessment would provide the basis for negotiations with the manufacturer. Future prices could rise and decline based on inflation, how well the drug continues to perform in real-world use, and the introduction of related products. It would make sense to create such an organization within the U.S. Department of Health and Human Services.
Another way of addressing prices during this period is to provide better information to prescribing physicians and patients. Currently, the vast majority of information that both doctors and their patients receive is a result of the billions of dollars that companies spend promoting their products. What is needed is more objective, noncommercial information about drug benefits, risks, and cost effectiveness. Policymakers should support independent programs that generate unbiased information about evidence-based management of disease and disseminate this educational information to physicians. This can translate into more cost-effective prescribing.
Transition to a competitive market period
Because the only type of competition that consistently and substantially lowers drug prices comes from the introduction of interchangeable, FDA-approved generic drugs, brand-name manufacturers often employ strategies to extend market exclusivity periods. Companies exploit ambiguities in the Patent Act to obtain dozens of “secondary” or “tertiary” patents on peripheral aspects of their approved brand-name drugs. The subjects of these follow-on patents can include anything from the medicine’s coating or a change in its delivery from a pill to a capsule, to using a new device such as an injectable pen. Incremental changes are commonly covered by these patents, but they often do not provide advancements in drug efficacy, safety, or convenience that are commensurate to the higher prices being charged.
In addition, manufacturers have used various other strategies to prevent the timely entry of generic drugs. These have included filing citizen petitions with the Food and Drug Administration raising frivolous concerns about the interchangeability of a potential generic, restricting supplies of their product for generic manufacturers to use in the bioequivalence studies needed to prove that a generic matches the original drug, and entering into settlements with generic manufacturers to drop patent challenges and delay their plans to market a competing generic product.
Policies to make those strategies more difficult or impossible to carry out can speed the introduction of competition and thus price reductions. A number of bills have been considered in Congress that address generic-delaying strategies in a piecemeal way. And in 2019, Congress passed and President Donald Trump signed into law bipartisan legislation intended to stop the practice by brand-name drugmakers of keeping supplies of medications out of the hands of generic manufacturers to prevent them from creating competing products.
Other measures Congress could take include requiring greater disclosure of a product’s patents (particularly for biologic drugs), preventing abuses of the Food and Drug Administration’s petition process, addressing remaining problematic settlement agreements with generic manufacturers, and giving the Food and Drug Administration greater authority to approve as interchangeable generics that are slightly different from the original drug if the differences are not clinically relevant. More frequent use of the Patent Trial and Appeals Board’s administrative patent review process—such as automatic review at the time any drug patent is FDA-listed—also could help weed out insufficiently innovative patents.
Another policy solution that Congress could enact is to restrict a brand-name drug’s market exclusivity period to a particular time period and not permit secondary or tertiary patents—or any of the other strategies—from being able to block FDA approval of a generic version. Manufacturers could be restricted to the single patent for which they seek and receive patent term restoration (a period of up to 5 years to account for time spent in clinical trials and FDA review), plus the 6-month patent extension manufacturers receive for testing their drugs on children. At the end of this period, generics would be permitted to enter, no matter what other patents have been obtained.
Multisource generic drug period
After a drug has lost exclusivity protection, prices may not fall if there are not enough generic manufacturers in the market, and other market conditions might also work against price declines. The Food and Drug Administration has deployed user fees, which began in 2012, to make substantial reductions in previous delays in the approval process for generic drugs. More resources must be invested at the agency to ensure that there are no unnecessary delays in generic drug approval, as well as in providing guidance on the types of studies generic manufacturers need to complete to receive FDA approval.
There are also some instances of older pharmaceuticals that no longer have exclusivity but for which not enough generic manufacturers have entered the market to reduce price substantially. In these cases, importation is a possible solution. This would involve setting up a regulatory system that would allow mutual recognition between the Food and Drug Administration and similar regulatory systems in Europe and in Canada, Australia, and Japan.
Another solution would be to authorize government manufacturing of generic drugs. Some private organizations and other nonprofit drug manufacturers have emerged in recent years, and a government-run manufacturing plant could ensure a continued supply of off-patent products that for-profit generic manufacturers have lost interest in producing. This idea has been proposed at both the national and state levels.
Conclusion
The pricing of remdesivir is emblematic of problems with the current process of developing and marketing prescription drugs in the United States. The policies described in this issue brief won’t discourage innovation, and they won’t reduce the availability of lifesaving and life-extending drugs—quite the opposite. They will turn the attention of successful companies away from efforts to create incrementally innovative products or to extend patent protection for their existing money-making products and toward the development of new ones. And they will relieve the financial burden that high prices place on consumers, businesses, and governments.





